
Edition 4- November 1997
DNA sequencing: A powerful new tool for the classification and identification of bacteria and fungi
Dr Wendy Untereiner (BCCM™/MUCL) and Dr Marc Vancanneyt (BCCM™/LMG)
Edition 4- November 1997 |
DNA sequencing: A powerful new tool for the classification and identification of bacteria and fungiDr Wendy Untereiner (BCCM™/MUCL) and Dr Marc Vancanneyt (BCCM™/LMG) |

DNA sequencing, an increasingly important part of BCCM™'s activities. |
Classification and nomenclature systems of all organisms are historically based on observable, morphological characteristics. Modern plant taxonomists, for example, employ many of the physically manifest or phenotypic characteristics used by their 18th century counterparts to discriminate between, for instance, birch trees and chrysanthemums. The same principle has been applied to naming species and underlies our understanding of how organisms are related to one another. We recognise, for instance, that house cats more closely resemble lions and tigers than dogs or polar bears: we anticipate that morphological similarities reflect close relationships between members of the cat family.
Systematists are aware, however, that phenotype may not accurately reflect true relationships or indicate evolution from a recent, common ancestor. Indeed, organisms can be remarkably similar phenotypically, simply because they occupy similar habitats or utilise the same resources. For instance, a number of animals have evolved wings, yet we hardly consider butterflies, birds, bats and pterodactyls to be closely related on the basis of the similarity of their forelimbs. This sharing of similar features among organisms with different phylogenetic histories is referred to as convergent evolution.
Although differentiating similarities due to common ancestry from those that arise in response to evolutionary pressures is a predicament for all taxonomists, convergence poses special problems for bacterial and fungal systematists. This is due to the fact that, of the few obvious or discriminating characters possessed by bacteria and fungi, many are the product of convergent evolution. For example, quite a number of bacteria, such as actinomycetes, exhibit filamentous growth. For years they were grouped with the fungi that also possessed this form of growth. Also, phenotypic characteristics are often unstable and may change or disappear within a few generations. Growth form in fungi is, for instance, dependent on environmental conditions and many groups are now recognised as possessing both filamentous and yeast-like phases. The consequence of using convergent or unstable taxonomic characteristics has resulted in the misclassification of bacteria and fungi, and a reliance on features that are inadequate to identify these organisms accurately.
The phenotypic approach is obviously also insufficient when studying bacteria and fungi that do not grow in culture. It is estimated that up to 95% of microorganisms are unculturable and as such, represent a vast, invisible assemblage that will remain unclassifiable as long as taxonomists rely on phenotypic characteristics alone.
 |
A more recent approach to the classification and identification of microorganisms involves the comparison of genotypic or genetic characteristics. Classical genotypic methods used in microbial systematics over the past thirty years include the determination of DNA base ratio (moles percent guanosine plus cytosine) and DNA-DNA hybridisation studies. The latter method has played a major role in the reclassification of bacteria and yeasts.
One of the groups of genes most frequently targeted for evolutionary relationship studies are those that code for ribosomal RNAs (rRNAs), or the molecules that constitute the core of the ribosome. These genes, known as ribosomal rDNAs, are useful because they include both slowly evolving or conserved regions as well as rapidly evolving or variable regions. They can thus be used to study the evolutionary divergence between distantly related organisms as well as closely related species. The development of methods for the indirect comparison of molecules enabled microbiologists to focus on comparative studies of rRNAs. For example, the hybridisation techniques pioneered by former BCCM™/LMG Director Prof. em. J. De Ley revealed natural relationships within a number of bacterial lineages.
Other approaches used to estimate genetic variation involve the use of electrophoresis, or the separation of macromolecules on the basis of differences in mobility due to their size, conformation and net charge. This methodology, which was first employed for the comparison of proteins (isozymes), has also been applied to detect variation in the pattern of randomly amplified polymorphic DNAs (RAPDs) and the fragments resulting from the digestion of genomic or amplified DNA with restriction enzymes (RFLPs). These techniques are highly discriminatory and are useful in determining infraspecific relationships. However, the most powerful electrophoretic tool involves the direct comparison of the sequence of nucleotides subunits of DNA. This approach has developed rapidly over the last decade and is widely employed in the characterisation and identification of organisms, as well as for inferring their phylogenetic or genealogical histories.
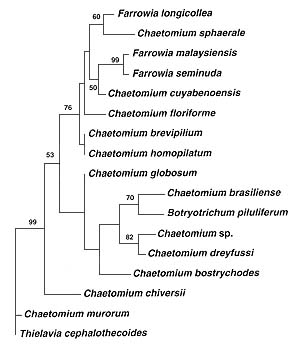 Phylogram of the relationships among members of the Chaetomiaceae based on parsimony analysis of 28S rDNA sequence data. |
Once a particular gene is selected for sequencing, many copies of this target DNA must be isolated. Although there are several strategies for obtaining large quantities of DNA, the most widely used is the polymerase chain reaction (PCR) method, a synthetic, cyclical process for amplifying DNA which exponentially increases the number of copies of the target sequence.
PCR products are sequenced using a procedure whereby the amplification product acts as a template for synthesising new strands of target DNA. However, because of the inclusion of specific chemical inhibitors in the reaction mixture, the newly synthesised DNA strands vary in length. Because they differ in size, these strands can be separated electrophoretically on polyacrylamide gels. In the past, these strands were labelled with radioisotopes and visualised by autoradiography (X-ray film). Modern sequencing procedures employ fluorescent chemical markers that are detected as the DNA fragments pass a stationary laser. The DNA sequence is recorded as a chromatogram where each "peak" corresponds to a single nucleotide. The chromatogram is recorded by a computer and converted automatically into data which can be compared with homological sequences from other individuals, species or genera.

Chromatogram from an automated DNA sequencer for fungal 28S rDNA |
For many years DNA sequencing was an expensive and labour intensive technique restricted to a few research laboratories, but now, the development of automated sequencing systems and computer programs for sequence data comparison has made this technology more cost efficient and more widely accessible. DNA sequencing is now considered the most powerful tool available to microbial systematists. Aside from its importance in the investigation of the evolutionary histories of microorganisms, sequence analysis is also a standard technique used in bacteria and yeast identification. Advances in sequencing technology have improved other methods for rapid microbial identification and typing, including in situ detection, by facilitating the design of nucleic acid probes used to detect bacteria and fungi in mixed microbial communities. This method has also greatly increased our understanding of the number and diversity of unculturable bacteria and fungi through studies which target species known only from their DNA sequences.
Bacterial and fungal DNA sequence data are collected by many research groups throughout the world and deposited in data banks accessible via the Internet (see 'Bioinformatics Corner'). Although only a few organisms' genetic complements are known in their entirety, portions of many thousands of species' genomes are available for comparison and homology searches in these sequence databases. These databases are expected to expand given the rapid advances in molecular technology, the decreasing cost of automated sequencing systems, and the recognition of the importance of DNA sequence data in organism classification and identification.
DNA Sequencing: a new BCCM™ serviceThe automated Applied Biosystems Prism™ DNA sequencers of BCCM™/LMG and BCCM™/MUCL are not only of exceptional value for internal research purposes, but have also proved to be irreplaceable in the frame of BCCM™'s quality services portfolio.
Indeed, for several months, both BCCM™-collections have been confronted with increasing numbers of requests for customised sequencing, coming from both academics and industrialists.
Firstly, there is a demand for sequencing of 16SrDNA or other target DNA (e.g. spacer ITS-regions for fungi and yeasts), with a view to characterise and/or identify a wide range of microorganisms. This service starts by amplifying target DNA, using the polymerase chain reaction (PCR). In the sequencing reactions, different primers are normally used, to assure maximum reliable results in the final consensus sequence.
Secondly - and this is with a view to unambiguous strain authentication - one is confronted with a growing need to determine, through contract research, strain-specific sequences in highly variable regions of microbial genomes. Indeed, the resulting 'sequence signatures' of, for example, proprietary production strains, reinforce major company policies in the fields of Quality Control or the protection of Intellectual Property Rights. In the latter respect it should be recalled that the Budapest Treaty strongly recommends the scientific description and/or proposed taxonomic designation of a micro-organism deposited for patent purposes.
Contact:
Dr W. Untereiner, BCCM™/MUCL
Ir B. Hoste, BCCM™/LMG |
 BCCM BCCMHome |
Contents Edition 4- November 1997 | Next Article Edition 4- November 1997 |